Heart failure is a chronic and lethal syndrome leads to a progressive decrease in cardiac function. In industrialized countries, it affects 10% of the population aged 65 and over. In the final stages of the disease cardiac output is no longer sufficient to match the oxygen and metabolic needs of the body, resulting in organ failure and death. A number of stresses and insults can trigger heart failure including long-term hypertension, myocardial infarction (MI), excess production of hormones and neurotransmitters, as well as exposure to drugs and toxicants. Ventricular cardiomyocytes initially adapt to the increased workload imposed by these cardiac insults by hypertrophying. While this compensatory process initially reduces the stress on ventricular walls and normalizes cardiac output, on the long term it predisposes to adverse ventricular remodeling associated with cardiomyocyte apoptosis, interstitial fibrosis, and impaired cardiac function.
Our laboratory investigates a range of focus areas, all of which center on understanding the molecular mechanisms governing heart dysfunction and repair. We have shown that the intracellular transduction events controlling these processes are regulated by scaffolding and anchoring proteins, which ensure coordination of signals in space and time. In particular, A-kinase anchoring proteins (AKAPs), assemble multifunctional signaling complexes that orchestrate and synchronize cellular processes associated with cardiac remodeling and protection.
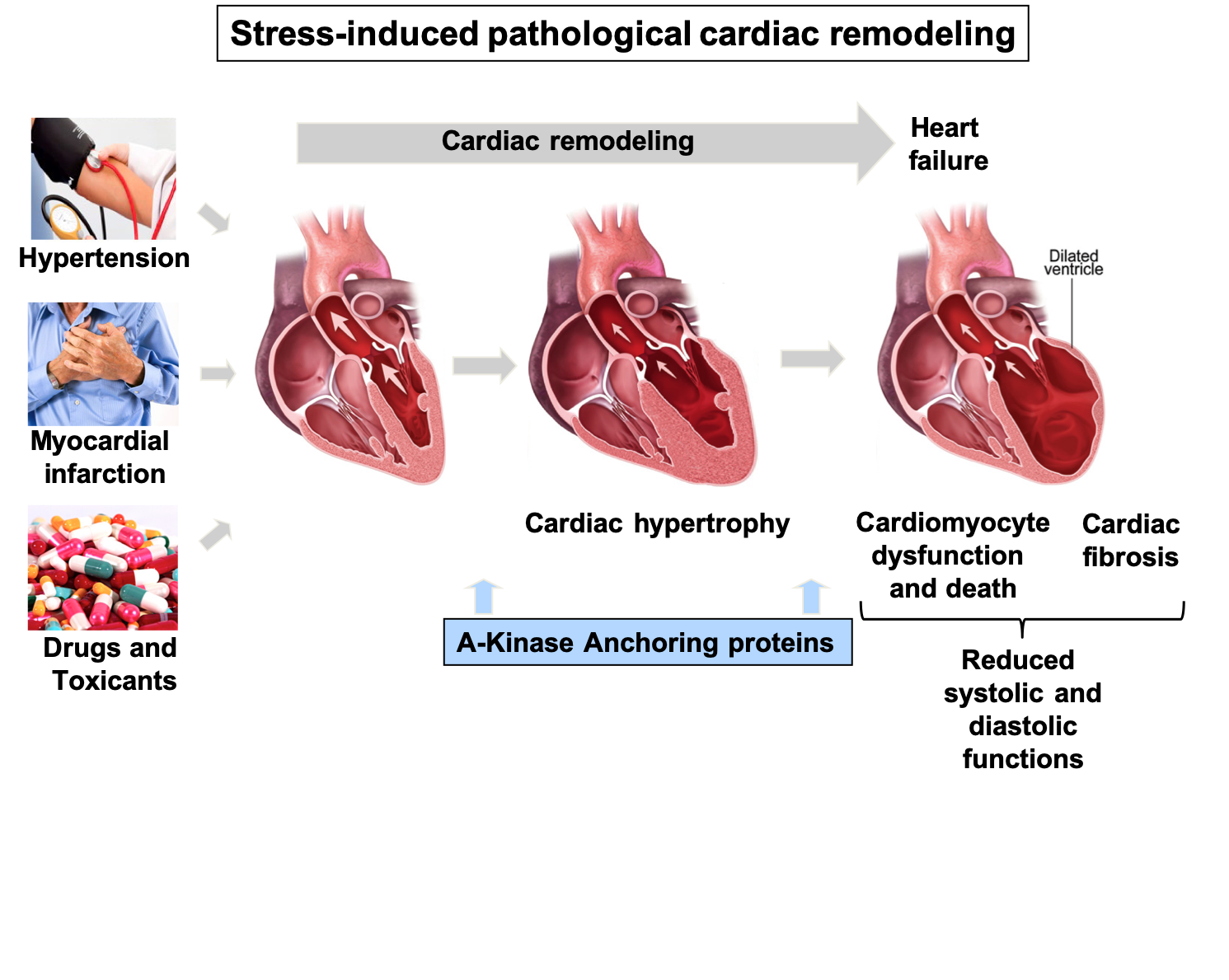
Schematic representation illustrating the cardiac remodeling process induced by various pathological stresses.
We are taking advantage of multidisciplinary approaches including genetically engineered mouse models, primary cultures of cardiomyocytes and fibroblasts, echocardiographic and life-imaging techniques, as well as state-of-the-art genomic and proteomic techniques to define the role of AKAP-based signaling complexes in regulating cardiomyocyte hypertrophy, inflammation and fibrosis in the stressed or injured heart. Similarly, we are examining how AKAPs regulate transcriptional responses involved in cardiomyocyte survival and cardiac protection.
1) The role of AKAPs in cardiac fibrosis
Stress-induced myocardial remodeling and fibrosis represent major causes of cardiac dysfunction in failing hearts. Accumulation of fibrotic tissue decreases myocardial compliance and affects electrical coupling between cardiomyocytes, reducing ventricular filling during diastole and causing arrhythmias, respectively.
In response to various cardiac stresses and insults such as MI and pressure overload, quiescent resident cardiac fibroblasts undergo transdifferentiation to myofibroblasts. These activated fibroblasts display increased proliferatory and migratory properties, as well as enhanced ECM synthetic capacity, which allows them to colonize and remodel the injured myocardium. We are investigating how specific alterations of AKAP-mediated signaling in cardiac fibroblast impacts the activation of fibrotic responses, the development of fibrosis and restores function in diseases hearts.
2) The role of AKAPs in cardiac protection
Preserving cardiomyocyte function and viability in stressed hearts is crucial for maintaing cardiac ouput and reducing the reducing the occurence of heart failure. We have shown that AKAPs expressed in cardiomyocytes assemble signaling units promoting survival pathways that favor compensatory hypertrophy, inhibit cardiomyocyte apoptosis and induce new vessel formation. We are currently interested in defining how these AKAP signaling units regulate the transcritional machinery and chromating remodeling processes associated with the activation of protective gene programs in the heart.
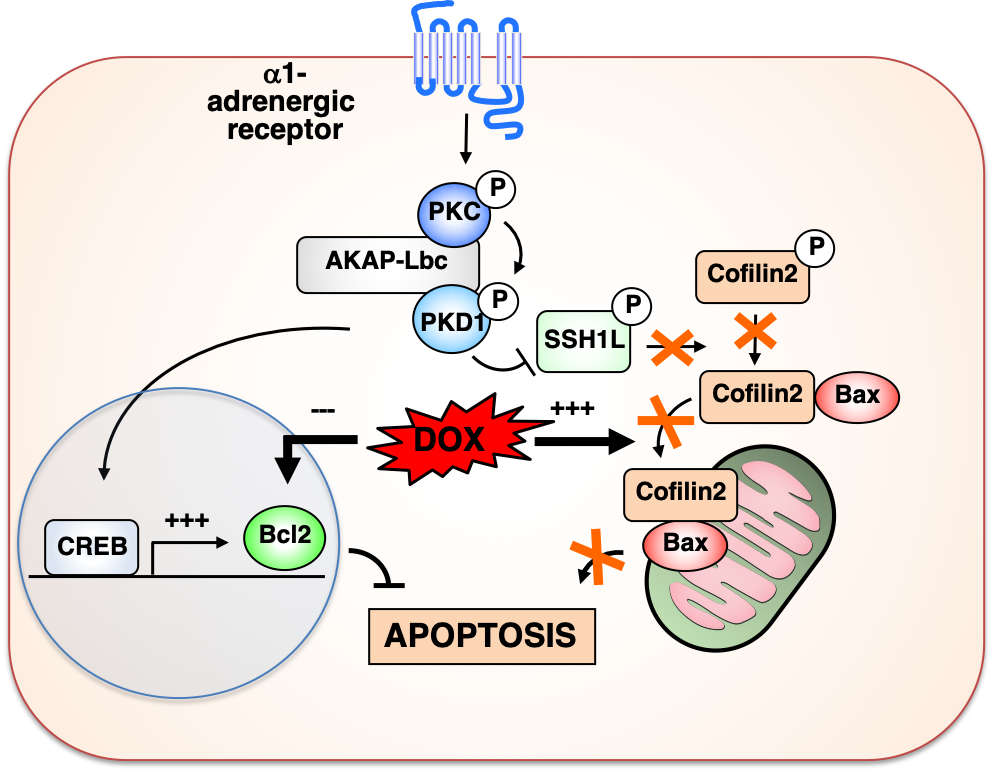
Model illustrating the role of the AKAP-Lbc/ protein kinase D1 signaling complex in mediating cardiomyocyte protection against doxorubicin-induced apoptosis. Doxorubicin (DOX) is a widely used anti-cancer agent known to promote cardiomyocyte death, severe cardiac dysfunction and heart failure. DOX-induced apoptosis of cardiomyocytes has been linked to the inhibition of the expression of anti-apoptotic proteins and to the ability of this anti-cancer drug to induce severe mitochondrial dysfunction. We have shown that AKAP-Lbc forms a complex with protein kinase D1 (PKD1), which mediates CREB-dependent rescue of anti-apoptotic Bcl2 expression in DOX-treated cardiomyocytes. In a parallel pathway, AKAP-Lbc facilitates α1-adrenergic receptor-induced activation of protein kinase D1, which in turn phosphorylates and inactivates the phosphatase SSH1L. This prevents cofilin2 dephosphorylation, which in turns inhibits DOX-induced translocation of cofilin2/Bax complex to mitochondria, thereby preventing cytochrome C release, mitochondrial dysfunction and apoptosis.
Dario Diviani, Associate Professor
 |
Dario Diviani received his PhD in 1998 from the University of Lausanne for research on alpha-1 adrenergic receptors performed with Prof. Susanna Cotecchia. Between 1998 and 2001, he performed a post doctoral training with Prof. John D. Scott at the Vollum Institute of the Oregon Health Sciences University, Portland, where he worked on the molecular mechanism controlling signaling specificity in heart cells. In 2001, he joined the Department of Pharmacology of the University of Lausanne, as an independent investigator. Since August 2018, he is the chairman of the Department of Pharmacology & Toxicology. |
Ivan Gautschi, Lab technician
 |
During 1987–1992, he worked in the group of Françoise Roch-Ramel on ion channel transport by using isolated perfused nephrons. During 1992-2018, Ivan worked in the group of Laurent Schild, carrying out Two-Electrode Voltage-Clamp experiments mostly with Epithelial Sodium Channel (ENaC). Since 2018, Ivan has been working in the lab of Dario Diviani where is applying lentivirus-mediated gene silencing technology to the study of cardiac fibroblast function. |
Marion Delaunay, PhD student
 |
Marion obtained her Bachelor in Biology at the University of Tours and her Master in Molecular and Cellular Biology at the University of Lyon. She performed her first year of Master’s internship in the lab of Prof. Kathrin Gieseler at the Neuromyogene Institute (Lyon) where she studied the role of a pro-apoptotic protein in Duchenne Muscular Dystrophy. During her Master’ thesis, she worked on directed manipulation of postnatal brain germinal activity with bioactive small molecules under the supervision of O. Raineteau at the Stem Cell and Brain Research Institute (Lyon). Afterwards she joined the group of Prof. Dario Diviani as a PhD student in March 2018 and is presently investigating the implication of A-Kinase Anchoring Proteins in stress-induced cardiac fibrosis. |
Simon Kaiser, PhD student
 |
Simon obtained his Bachelor in Biology at the University of Neuchâtel and his master in Medical Biology at the University of Lausanne. During his pre-master project, he worked on the implication of the cystathionine-γ-lyase in the protection of endothelial cells against palmitate-induced apoptosis under the supervision of Florent Allagnat. Then, he joined Dario Diviani’s lab for his master thesis. He worked on the role of AKAP2 in prostate cancer development and more precisely it’s implication in tumor’s migration. Thereafter he stayed in Dario Diviani’s lab as PhD student in March 2019 and continued working on the role of AKAP2 in prostate cancer development. |
Greta Scherler, PhD student
 |
Greta obtained her Bachelor in Biology at the University of Lausanne, where she is now performing a Master in Medical Biology, with a specialization in pharmacology and toxicology. She performed her pre-master project in the laboratory of Marie-Christine Broillet in the Department of biomedical sciences, studying the role of the olfactory marker protein (OMP) on the activation of olfactory sensory neurons in mice. Then, she joined Dario Diviani’s Lab to perform her Master thesis, investigating the implication of A kinase anchoring proteins in cardiac fibrosis, focusing on their role in collagen secretion in cardiac myofibroblasts. |
Simone Spinozzi, Post-doc
 |
After graduating with a Bachelor’s Degree in Biotechnologies and a Master’s Degree in Medical Biotechnologies at the University of Siena in Italy, Simone started a PhD in the group of Prof. Vincenzo Sorrentino, where he was studying the molecular physiopathology of myopathies caused by mutations in triadic and sarcomeric proteins. Once he obtained his Doctoral Degree in Molecular Medicine, he joined Prof. Ju Chen’s Laboratory at the University of California San Diego in USA as a Postdoctoral Researcher, to study the molecular mechanisms underlying genetic mutations that alter cardiac EC-Coupling and catecholaminergic signaling. Then, he moved back to Europe as a Research Associate at the Généthon Institute in France, where he was developing AAV-based gene therapies for cardiac and musculoskeletal genetic disorders. In 2022, Simone joined the group of Prof. Dario Diviani here at UNIL, where he is studying the role of A kinase anchoring proteins (AKAPs) in cardiac myofibroblast-mediated myocardial repair and remodeling. |